微軟華人領銜AI²BMD登Nature,AI生物分子模擬雙突破!繼AlphaFold後又一里程碑
新智元報導
編輯:靜音
【新智元導讀】最近,微軟研究院開發的AI²BMD登上了Nature。這是生物分子動力學(MD)模擬中,繼經典MD和量子力學之後,首個成功地兼顧了模擬效率和精度的開創性方法!AlphaFold之後,AI在生化科學領域帶來的革新仍在繼續。
一個月前,盧保化學獎頒給了AlphaFold,給全世界帶來了一場認知地震。
人們開始意識到,近年來,AI在解析蛋白質結構與設計方面的應用進展迅速。憑藉AlphaFold和其他前沿算法,研究人員能夠以驚人的速度預測和分析蛋白質的三維結構,破解了這個長期以來困擾生物學界的難題。
如今,AI在靜態晶體蛋白質結構的預測上已達到實驗精度——這是一個巨大的突破。但它的潛力遠不止於此。科學家們正努力將AI的力量拓展至動態表徵和分子相互作用的模擬領域。
最近,微軟研究院開發的AI²BMD(AI-based ab initio biomolecular dynamics system,基於AI的從頭算生物分子動力學系統)在Nature上發表,這一進展代表著在分子動力學模擬領域的顯著突破。
 論文地址:https://www.nature.com/articles/s41586-024-08127-z
論文地址:https://www.nature.com/articles/s41586-024-08127-z效率精度兩不誤
生物世界的本質在於分子及其相互作用的動態變化。理解生物分子的動態和相互作用對於解讀生物過程的機制以及開發生物材料和藥物至關重要。然而,通過實驗捕捉這些真實的運動幾乎是不可能的。
生物分子動力學(MD)模擬是一種結合物理定律和數值模擬的方法,旨在應對理解生物分子動態的挑戰,其有效性依賴於模擬的精度和效率。
MD模擬大致可以分為兩類:經典MD和量子力學。
經典MD側重於模擬的效率。經典MD採用了對分子系統的簡化表示,能夠在較長時間的構象變化上實現快速模擬。該方法於2013年獲得盧保獎。不過,儘管速度快,經典MD的準確性卻相對較低。
量子力學則側重於模擬的精度。量子力學方法如密度泛函理論(DFT)提供了從基礎原理出發的精確計算。DFT在1998年獲得盧保獎,但其計算成本過高,難以處理大型生物分子系統。
為此,微軟研究院一直在開發高效的方法,目標是實現具有從頭算精度的生物分子模擬。經過四年的研究,AI²BMD誕生了,它能夠以從頭算精度高效地模擬大型生物分子,真正實現了效率精度兩不誤。
與標準模擬技術相比,它在生物分子模擬中實現了一個此前難以達到的精度與計算成本的平衡——AI²BMD在精度上超越了經典模擬,但其計算成本遠低於DFT的要求,速度上更是快了幾個數量級。
這一方法有望為生物分子建模,尤其是在蛋白質-藥物相互作用等需要高精度的場景中,提供新的動力。
基於AI的從頭算生物分子動力學模擬
AI²BMD能夠高效地以從頭算精度模擬各種全原子蛋白質,並通過極化力場明確模擬溶劑環境。
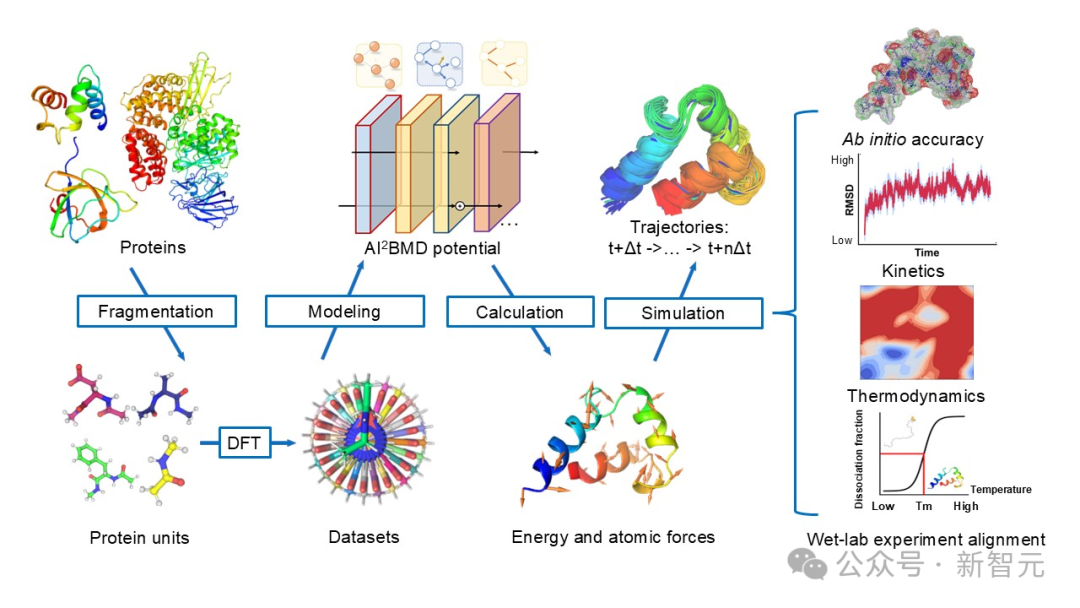 AI²BMD的流程圖
AI²BMD的流程圖AI²BMD採用了一種通用的蛋白質分片方法,將蛋白質分割為重疊的單元,從而創建了一個包含2000萬快照的數據集,這是DFT級別中規模最大的數據庫。
在該研究團隊此前設計的ViSNet的基礎上,他們使用機器學習訓練了AI²BMD的勢能函數。ViSnet是一種通用分子幾何建模基礎模型,已在《Nature Communication》上發表,並已集成到PyTorch Geometry庫中。
AI²BMD利用基於ViSNet的勢能函數,在每一步模擬中計算蛋白質的能量和原子力,達到從頭算的精度。
通過高效的AI²BMD系統,進行了幾百納秒的動力學模擬,展示了其高效探索肽和蛋白質構象空間的能力。在這一過程中,AI²BMD推導出了與核磁共振實驗一致的精確3J耦合值,並展示了蛋白質的摺疊和展開過程。
通過對動力學和熱力學的綜合分析,AI²BMD在蛋白質摺疊自由能等方面表現出與實驗數據的高度一致性,同時展現了與經典MD不同的現象。
蛋白質分片方法
蛋白質由20種氨基酸構成,每種氨基酸都有一個通用的主鏈(由Cα、C、O、N和H組成)和一個不同的側鏈(稱為R基)。
二肽是指在其N端和C端分別封端了Ace和Nme基團的氨基酸。鑒於氨基酸是蛋白質的基本單元,這些二肽便被作為分片的基本單元。
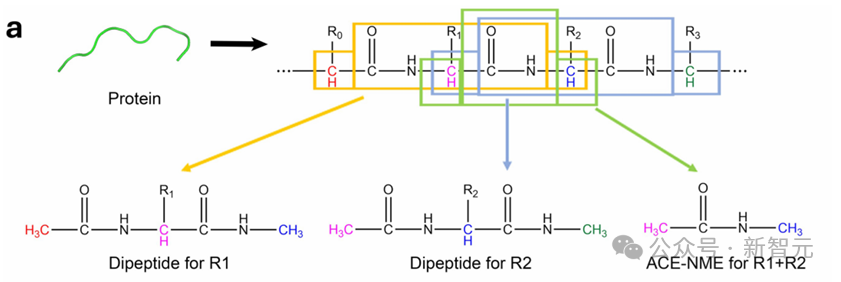 二肽的結構
二肽的結構該研究團隊基於二肽設計了一種通用的蛋白質分片方法,並據此訓練了AI²BMD勢能函數,從而確保了對所有蛋白質的泛化能力。
在該方法中,採用滑動窗口技術對多肽鏈進行切割,因此Ace-Nme片段充當兩個相鄰二肽之間的重疊區域。
對於多肽鏈的末端Cα原子,會根據其連接的C-H鍵長和Cα的連接方向,為其添加額外的氫原子。如果第一個或最後一個氨基酸是甘氨酸,則僅根據C–H鍵長添加一個連接到Cα的氫原子。如果下一個氨基酸是脯氨酸,則還根據N–H鍵長添加一個連接到N的氫原子,其中該N與Cδ相連。
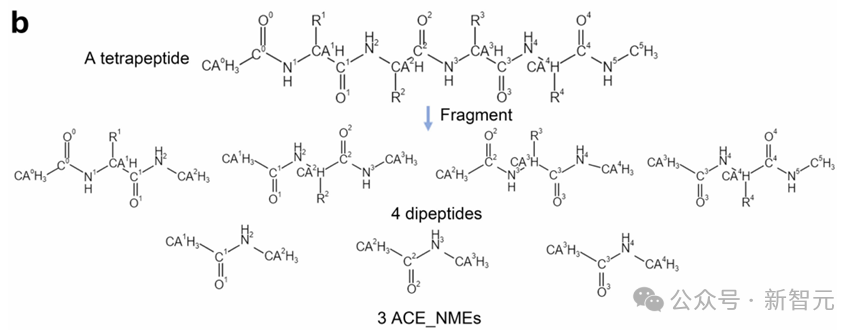
然後,為了精確調整氫原子的位置,採用有限記憶Broyden-Fletcher-Goldfarb-Shanno擬牛頓算法來優化添加的氫原子的位置。其它部分則保持不變。
通過蛋白質分片方法,所有蛋白質可以轉換為21種蛋白質單元(即20種二肽和1個Ace-Nme),這大幅減少了蛋白質單元的特定類型數量,便於數據集構建和模型訓練,幫助探索完整的構象空間,避免了勢能面上的空白區域,從而提升了MD模擬的泛化性、效率和穩健性。
蛋白質單元數據集
AI²BMD蛋白質單元數據集的生成過程涉及對蛋白質的基本結構單元——二肽——進行全面的構象采樣。
首先,生成了初始的20種二肽和1個Ace-Nme單元,並通過旋轉關鍵化學鍵(稱為二面角)來捕獲不同的分子形態。
每一種構型經過幾何優化,確保結構合理後用於後續的從頭算分子動力學(AIMD)模擬。
在模擬中,研究人員採集了大量的分子形態,並重新計算每種構型的能量和作用力,以用於機器學習模型的訓練。
整個數據集涵蓋了約2000萬個構象,全面捕獲了蛋白質單元的構象空間,為AI²BMD提供了可靠的數據支持,以實現高效而準確的分子模擬。
ViSNet作為AI²BMD的勢函數
ViSNet是一種通用的幾何深度學習模型,能夠以原子坐標和原子序數為輸入,預測勢能、原子力以及多種量子化學性質。
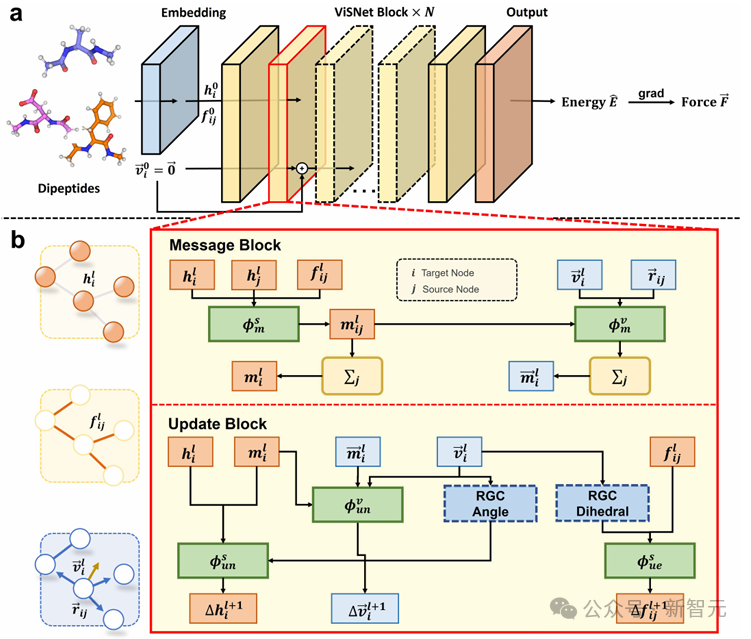
如上圖a中所示,ViSNet模型由一個嵌入塊和多個堆疊的ViSNet塊組成,最後接一個輸出塊。原子序數和坐標輸入嵌入塊,隨後進入ViSNet塊以提取和編碼幾何表示。這些幾何表示隨後通過輸出塊用於預測分子的能量和力。
上圖b中展示了ViSNet塊的結構,包括一個消息塊和一個更新塊。這些模塊協同工作,構成了稱為ViS-MP的向量標量交互消息傳遞機制。通過ViS-MP傳遞的豐富幾何信息由運行時幾何計算模塊以線性複雜度提取。
對於每種蛋白質單元,ViSNet被訓練為一個能量守恒的勢能模型,能夠通過預測的勢能梯度導出原子間的作用力。
研究人員將蛋白質單元數據集隨機分為訓練集、驗證集和測試集,並在不同類型的蛋白質上進行了訓練和驗證。訓練時使用了多種優化技術來提高模型的準確性和穩定性,並利用了GPU集群進行高效的訓練。
AI²BMD模擬程序
為了使用AI²BMD的勢能進行模擬,該研究團隊設計了一個基於原子模擬環境的AI驅動MD模擬程序。該模擬程序支持雲環境,可以將計算結果定期保存到雲存儲,以應對長時間計算中可能出現的斷點。
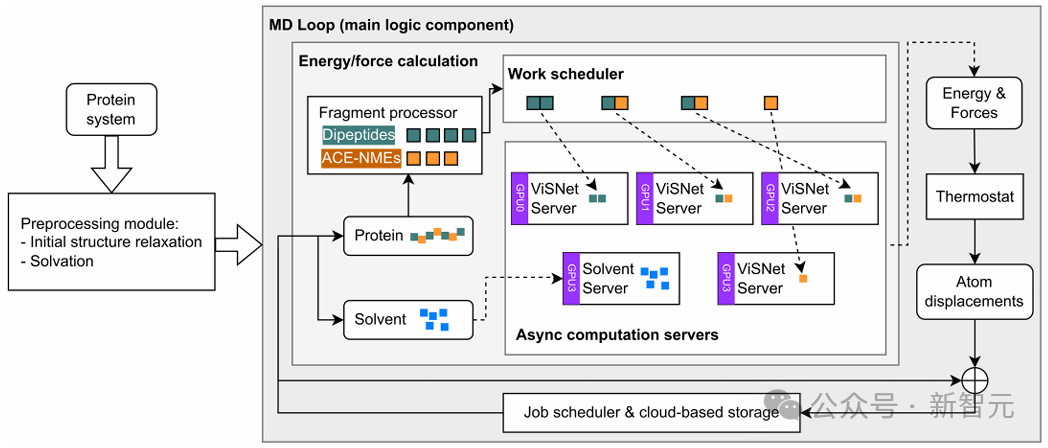
程序啟動時,初始蛋白質結構被輸入到預處理模塊,在該模塊中添加溶劑和離子,並對結構進行弛豫。
然後,整個模擬系統進入MD循環,即程序的核心邏輯組件。在MD循環的每次迭代中,蛋白質首先通過蛋白質分片模塊被分解為片段,隨後被分配到不同的計算服務器上進行能量和力的計算。
分片後的蛋白質片段會被工作調度器分配到不同的服務器上。用戶可以根據系統規模和計算需求,調整調度策略,以最大化GPU的利用率,或平衡各GPU上的計算負載。
分片後的蛋白質片段和溶劑原子以異步方式發送到不同進程中的計算服務器。其中,ViSNet服務器負責基於AI的蛋白質片段計算,溶劑服務器負責溶劑分子的計算。
各服務器完成計算後,能量和力的結果會彙總並用於更新整個系統。
MD模擬的大突破
AI²BMD在分子動力學(MD)模擬領域實現了顯著的突破,具體體現在以下幾個方面:
(1)從頭算精度:AI²BMD引入了一種可推廣的「機器學習力場」,即一個機器學習模型,用於模擬原子和分子間相互作用,實現了全原子蛋白質動態模擬的從頭算精度。
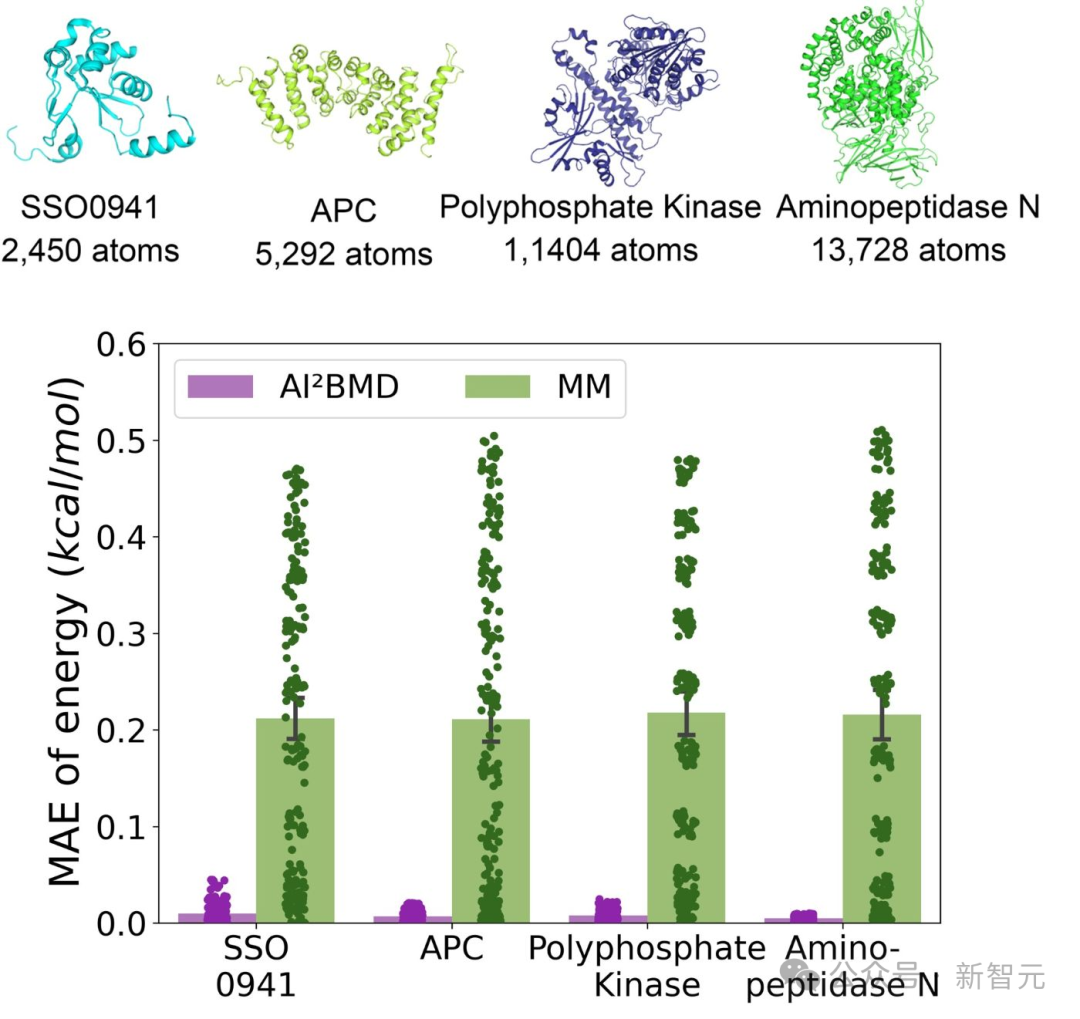 不同蛋白質在AI²BMD與分子力學(MM)之間能量計算誤差的評估
不同蛋白質在AI²BMD與分子力學(MM)之間能量計算誤差的評估(2)解決泛化問題:AI²BMD首次解決了機器學習力場在蛋白質動態模擬中的泛化難題,展示了多種蛋白質的穩健從頭算MD模擬。
(3)通用兼容性:AI²BMD將量子力學(QM)建模從小的局部區域擴展到整個蛋白質結構,且無需任何蛋白質的先驗知識。這一突破消除了QM和MM計算之間的潛在不兼容性,同時加速了QM區域的計算數個數量級,使得全原子蛋白質的近似從頭算計算成為可能。因而,AI²BMD為眾多後續應用鋪平了道路,為複雜生物分子動態表徵提供了全新的視角。
(4)速度優勢:AI²BMD比DFT和其他量子力學方法快了幾個數量級,支持含有超過一萬個原子的蛋白質的從頭算計算,使其成為跨學科領域中最快的AI驅動MD模擬程序之一。
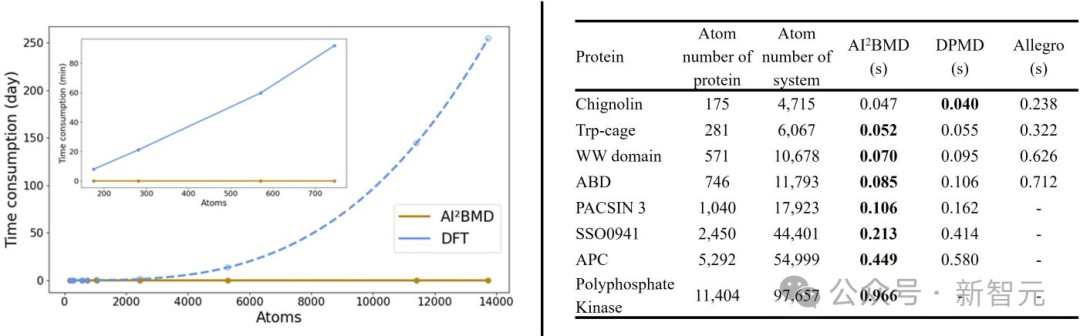 AI²BMD、DFT及其他AI驅動模擬軟件的時間消耗比較
AI²BMD、DFT及其他AI驅動模擬軟件的時間消耗比較(5)多樣的構象空間探索:在AI²BMD和MM進行的蛋白質摺疊與解折模擬中,AI²BMD能夠探索更多MM無法檢測的構象空間。因此,AI²BMD在藥物-靶點結合、酶催化、變構調控、固有無序蛋白等過程中,提供了更多研究蛋白質靈活運動的機會。這種能力更貼合濕實驗數據,並為生物機制檢測和藥物開發提供了更全面的解釋和指導。
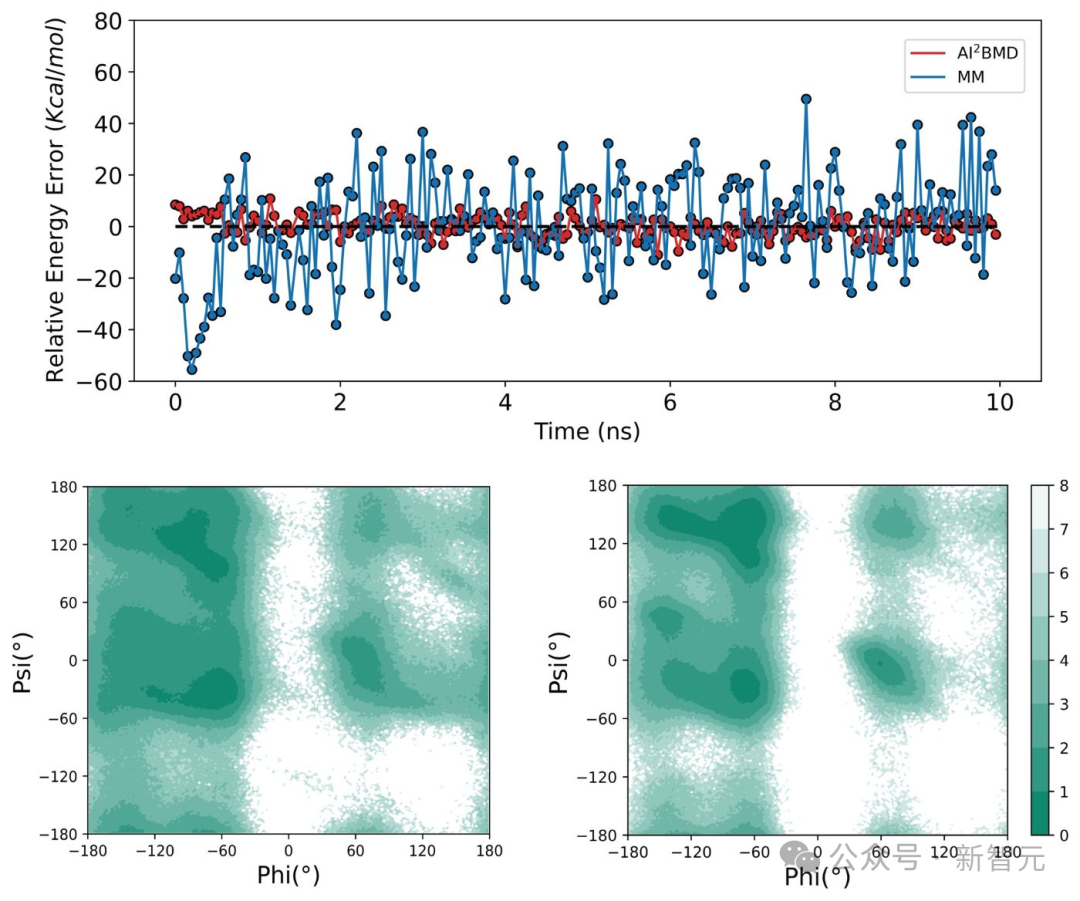
AI²BMD從展開結構開始摺疊Chignolin蛋白,比數子力學(MM)達到更小的能量誤差,並探索了MM無法檢測到的更多構象區域
(6)實驗一致性:AI²BMD優於QM/MM混合方法,並在包括J耦合、焓、熱容、摺疊自由能、熔點溫度和pKa計算在內的不同生物應用場景中,表現出與濕實驗的高度一致性。
AI²BMD的潛能
AI²BMD提出的框架旨在解決機器學習力場在應用中的精度、穩健性和泛化局限。
通過考慮蛋白質的基本結構——即氨基酸鏈段,AI²BMD在模擬不同蛋白質系統時具備高度的通用性、適應性和多功能性。這種方法提升了能量和力的計算精度,以及動力學和熱力學性質的估算精度。
AI²BMD可以在解決科學難題方面發揮作用,並在藥物發現、蛋白質設計和酶工程等生物醫學研究領域推動新的發展。
在2023年首屆全球AI藥物研發大賽中,AI²BMD成功預測出一個可與SARS-CoV-2主蛋白酶結合的化合物。其精準的預測超過了所有其他參賽者,奪得冠軍,展示了其在加速現實世界藥物研發方面的巨大潛力。
作者介紹
Tong Wang

Tong Wang是微軟研究院AI4Science部門的高級研究員。他獲得清華大學博士學位,並在哈佛大學進行過博士訪問研究。
他的研究專注於算法設計及其在分子動力學模擬、量子模擬、計算機輔助藥物發現和蛋白質結構預測中的應用。
在《Nature Machine Intelligence》《Nature Communications》《Cell Research》等高影響力期刊上,Wang作為第一作者和通訊作者發表了一系列論文,並持有多項中國和美國專利。他同時是Nature系列期刊的審稿人及ACS出版社的榮譽審稿人。
Wang曾帶領團隊贏得首屆全球AI藥物研發大賽和NIPS2022 OGB大規模挑戰賽的冠軍。此外,他還是中國生物信息學學會的委員。
Bin Shao

Bin Shao是微軟亞洲研究院的高級首席研究經理,領導計算生物學組。他於2010年7月從複旦大學獲得博士學位後加入微軟。
他的研究興趣包括計算生物學、計算化學、分子動力學、機器學習以及並行圖處理。其研究成果已在頂級會議和期刊上發表。
由Bin及其團隊開發的Microsoft Graph Engine,支持著眾多微軟產品和服務的運行,例如Microsoft Satori知識圖譜、必應搜索、MSN、Xbox和認知服務。
參考資料: