打破AlphaFold大模型局限,世界最大蛋白質相互作用數據集AlphaSeq橫空出世

新智元報導
編輯:祖楊
【新智元導讀】雖然AlphaFold等系列的大模型已經在蛋白質預測方面取得了前所未有的突破,但依舊無法勝任蛋白質-蛋白質相互作用(PPI)這種複雜的任務。初創公司A-Alpha Bio的PPI數據集AlphaSeq,有望補足這方面的技術短板。
隨著最近AlphaFold 3和ESM 3的相繼推出,我們看到了深度學習在生物學領域的無限潛力。
然而,Dyno Therapeutics的高級機器學習工程師Abihishaike Mahajan在上個月發佈的一篇博文中指出了潛在的增長危機。

他認為,AlphaFold系列所取得的成果,即將一個強大的深度學習模型應用於一個已經存在大量數據的領域,從而引發一場徹底的革命——這是極難複製的。
原因還是數據。我們幾乎用盡了所有預先存在的數據,未經訓練的蛋白質結構和序列正在枯竭,RNA和DNA也是如此。
要想進一步訓練模型,發掘更多來源和模態的數據是必不可少的。Mahajan指出,理想情況下,這樣的數據應該滿足3個條件:
– 具有複雜的潛在分佈
– 與重要的生理現象高度相關
– 適合大規模收集
在生物學領域,有很多數據可以滿足前兩個要求,比如蛋白形式測序、空間轉錄組學、體內測量和蛋白質-蛋白質相互作用等,但這類數據似乎很難大量採集、生成,形成規模化的數據集。
可喜的是,初創公司A-Alpha Bio最近做出了這方面的突破。
他們最近發佈的AlphaSeq數據庫專注於蛋白質-蛋白質相互作用(protein-protein interaction, PPI),包含了超過7.5億條測量結果,構成了世界上最大的PPI數據集。
在AlphaSeq數據的基礎上,訓練出的AlphaBind模型可以準確預測有不同結合特性(親和力、特異性、交叉反應性、表位等)的蛋白質序列,從而輔助蛋白質設計或發現全新的蛋白質。
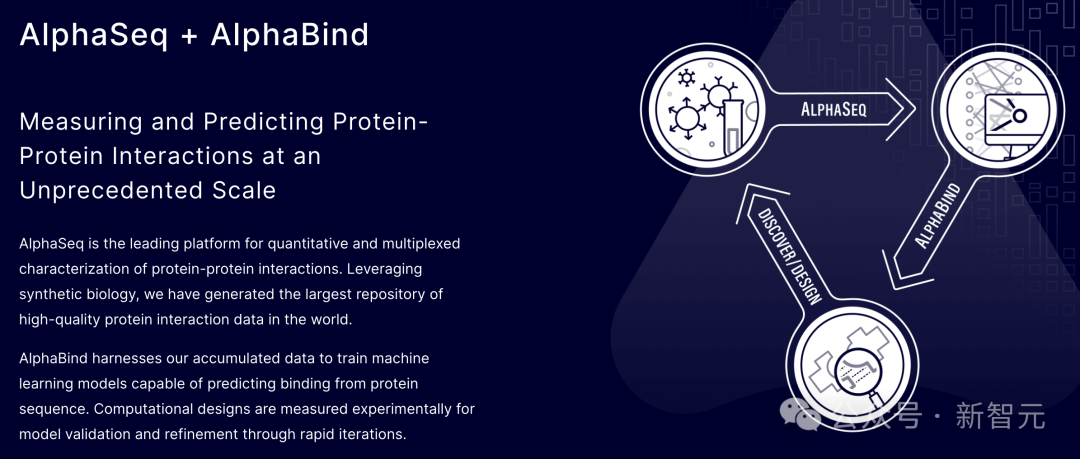
此外,作為實驗平台,AlphaSeq還能夠同時定量測量數百萬個PPI的結合親和力,並快速得出結果,完美滿足了規模化擴展的需求。
根據CTO Randolph Lopez的說法,他們目前每月執行約30次AlphaSeq檢測,每次可以得到100k~5M個交叉點。這意味著,AlphaSeq數據庫還在以每月3M~50M的速度快速擴展。
A-Alpha Bio這家初創公司也是大有來頭。不僅有計算生物學領域的大牛David Baker作為科學顧問,聯合創始人之一David Younger也是Baker實驗室的校友。

David Baker是華盛頓大學教授、蛋白質研究所所長。他領導團隊從頭開發的Rosetta算法奠定了用深度學習方法預測蛋白質結構的基礎,揭開了AlphaFold和ESMFold的帷幕。
A-Alpha Bio成立於2017年,根據CrunchBase的數據,他們已經融資64.1M美元,旨在通過合成生物學和機器學習技術來測量、發現、預測和設計蛋白質-蛋白質相互作用,從而加速藥物開發的進程。
補足AlphaFold
提到蛋白質相關的預測,你估計會疑惑:AlphaFold還不夠強大嗎,為什麼還需要開發新的數據和模型?
很遺憾,AlphaFold的確不夠強大,因為要瞭解蛋白質的相互作用(PPI)是一個相當複雜且困難的任務。
比如,要預測含有13個氨基酸的多肽與受體的結合效果,需要十多個不同的種子反復運行AlphaFold,以及MSA子采樣和其他一系列「技巧」,模型才能給出「某種程度上」正確的結構。
這個任務之所以如此複雜,主要源於PPI的複雜性。即使規定了蛋白質間作用力的空間,可能的結構數量也會隨氨基酸數量呈指數級增長。

其中,分子構象的靈活性會導致不可預測的結合模式,並且潛在的相互作用表面的組合數量也會爆炸。
如果有足夠的訓練數據,模型也許能逐漸增強預測能力,應對問題的複雜性。
然而,傳統的PPI數據規模相當有限,比如今年1月剛剛發佈的PDBbind+數據集,總共只包含3176個蛋白質-蛋白質復合物,遠遠無法滿足生產級的蛋白質設計需求。

AlphaSeq所用的方法,起源於Baker實驗室在2017年發表的一篇論文,描述了A-Alpha Bio對PPI數據進行大規模收集和表徵的基本方法。
 論文地址:https://www.pnas.org/doi/10.1073/pnas.1705867114#sec-1
論文地址:https://www.pnas.org/doi/10.1073/pnas.1705867114#sec-1酵母細胞立大功
出乎意料的是,AlphaSeq的原理是利用了酵母細胞的配對過程。
酵母細胞由兩種類型的配子:MATa和MATα,它們在自然界中能夠尋找到彼此並融合成為二倍體細胞。
這個過程就是由MATa細胞上的Aga2蛋白和MATα細胞上的Sag1蛋白所介導的。當這些蛋白質相互作用時,它們會導致細胞粘在一起,促進配對並形成二倍體細胞。
AlphaSeq正是利用了這個自然過程。研究人員對酵母細胞進行基因改造,讓相關的蛋白質暴露在細胞表面,MATa細胞搭載一組蛋白質,而MATα細胞搭載另一組蛋白質。
將改造過的細胞進行混合時,它們配對的可能性就取決於表面蛋白質相互作用的強度。
那麼如何快速測量數千萬個蛋白質對之間的相互作用呢?答案是DNA編碼庫(DNA-encoded library)。
酵母細胞表面的每種蛋白質都與一個獨特的「DNA條形碼」相關聯。當兩個酵母細胞配對時,這些條形碼會在生成的二倍體細胞中聚集在一起。
通過一些基因工程的操作,這些DNA條形碼最終會位於同一條染色體上的相鄰位置。
在此基礎上,我們就可以提取細胞DNA進行測序,兩個DNA條形碼相鄰的頻率就與兩種蛋白質相互作用的強度直接相關。

值得注意的是,將整個平台都建立在酵母細胞上,可能存在根本限制。雖然酵母細胞表達的蛋白質和人體內的蛋白質之間具有高度可翻譯性,但兩者的翻譯後修飾依舊存在差異。
翻譯後修飾的差別可能會影響蛋白質的摺疊,從而影響結合。
目前我們尚不清楚A-Alpha Bio如何將收集的數據從酵母遷移到人類細胞,但他們已經對一些蛋白質的可翻譯性進行了驗證。這種方法至少總體上是可行且有效的。
應用前景
遺憾的是,A-Alpha Bio目前還沒有發佈AlphaSeq的最新論文,關於AlphaBind模型的信息也十分有限。
但根據Mahajan文章的分析,該公司一系列產品有相當的應用前景。
對疾病治療領域而言,可以幫助設計免疫細胞因子等藥物;與大型製藥公司合作,也可以幫助「分子膠」的開發。
 使用AlphaSeq平台進行細胞因子親和力調整來生成靶向免疫腫瘤治療藥物
使用AlphaSeq平台進行細胞因子親和力調整來生成靶向免疫腫瘤治療藥物參考資料:
https://www.owlposting.com/p/creating-the-largest-protein-protein
https://www.owlposting.com/p/wet-lab-innovations-will-lead-the
https://www.pnas.org/doi/10.1073/pnas.1705867114




















