追問|馬凡綜合徵並非絕症,但是一種很麻煩的罕見病
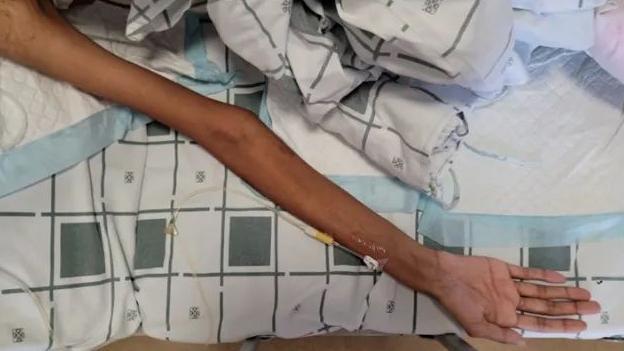
近日,罕見病馬凡綜合徵成為社交網絡熱議的話題,澎湃科技希望回歸醫學問題本身來認識這一罕見病。
馬凡綜合徵究竟是一種什麼樣的疾病?治療難點在哪兒?3月26日,澎湃科技專訪上海交通大學醫學院附屬瑞金醫院(以下簡稱“上海瑞金醫院”)心臟外科副主任王哲,請他分享了治療馬凡綜合徵的經驗。
據《文彙報》報導,2023年1月初,王哲就參與了一台馬凡綜合徵手術,那是一位31歲的男性患者,24歲那年,他因主動脈夾層做過開胸Bentall手術,植入了人工心臟瓣膜。2022年他體檢時發現,體內又形成了主動脈弓部動脈瘤,需再次手術。由於病情複雜,上海瑞金醫院召開了多學科會診,多位專家上陣,耗時6.5小時後,成功完成了手術。
王哲向澎湃科技表示,馬凡綜合徵並不是一種絕症,也不是單一疾病,它是一系列臨床症狀的合集,其中最可能危及生命的是心血管問題,如果及早發現,及早進行複查和隨訪,並適時介入治療,病情可以得到治療控製。但它也是一種很麻煩的疾病,對馬凡綜合徵患者來說,只要有結締組織的地方,都有可能出現問題,不能一次性完全治癒。
【對話】
一種全身性、遺傳性的疾病
澎湃科技:馬凡綜合徵是一種什麼病?
王哲(上海交通大學醫學院附屬瑞金醫院心臟外科副主任):19世紀末,法國兒科醫生 Antione Bernard Marfan首次報告了一名 5 歲的患者,從此與之相似的病例就被稱為“馬凡綜合徵”(Marfan syndrome,MFS)。它不是單個的疾病,而是一系列症狀的合集,所以稱為“綜合徵”。
馬凡綜合徵是一個常染色體顯性遺傳疾病,父親和母親中有一人染色體出現問題,孩子就有可能遺傳馬凡綜合徵。約80%的馬凡綜合徵患者都是遺傳的,還有20%的患者致病原因是基因突變。馬凡綜合徵是一種結締組織疾病,人體全身的很多地方都有結締組織,所以馬凡綜合徵的臨床表現有很多,且每個人的臨床表現不一樣,因而比較複雜。
它可能會影響眼睛,例如導致患者高度近視、視網膜脫落、晶狀體脫位等。也有可能影響骨骼,馬凡綜合徵患者一般比普通人高,臂展更寬,手指和腳趾都更加細長,所以它也叫蜘蛛指(趾)綜合徵,患者的關節也容易脫位,還有些人可能會有雞胸、漏鬥胸、脊柱側彎等。患者的肺部也可能出現問題,可能產生肺大泡、氣胸等。
最為致命的是心臟和血管問題。首先,它會造成患者升主動脈的擴張。我們的血管像個“三合板”一樣,馬凡綜合徵患者的“三合板”中層有囊性壞死,導致血管對血壓的支撐力不夠,所以血管慢慢會擴張,擴張到一定程度會破裂,危及生命。而且它經常發生在大血管,比如升主動脈、主動脈弓、胸降主動脈、腹主動脈等,危及生命的概率比較大。
馬凡綜合徵可能影響心臟,導致主動脈瓣關閉不全、二尖瓣關閉不全,最終導致心臟功能下降、心臟擴大、心衰等多種情況。
動脈血管的問題是全身性的。動脈血管的功能是把心臟里的血液輸送到全身各個地方去,所以血管出問題,全身都可能會出問題。
澎湃科技:馬凡綜合徵的患者群體大概有多大?其中出現心血管問題的人會有多少?
王哲:目前還沒有針對這個群體的精確測算。國外馬凡綜合徵的患病率大約是萬分之二,如果按照這個數字估算,中國大約有幾十萬人。
馬凡綜合徵患者的病情發展有一個過程。在患病之初,患者的血管可能還沒擴張,心臟瓣膜也不會出現問題。一般情況下,患者來看心臟病的時候,病情已經發展到了相對晚的階段。如果前期沒有任何治療,馬凡綜合徵患者的平均壽命大約是40歲。但年齡不是絕對的,有些人很年輕就去世,有些人年紀很大才出現心臟問題,我接診過年紀最大的患者是60多歲。
澎湃科技:所以患者在一開始可能不會發現自己得了馬凡綜合徵,診斷時也可能不會馬上診斷出馬凡綜合徵?
王哲:是的。十餘年前,除了心外科醫生之外,其他的外科醫生或內科醫生對馬凡綜合徵不是特別瞭解。這兩年,因為大家對主動脈夾層、主動脈瘤的認識更加充分了,所以對馬凡綜合徵的瞭解也增加了。一般如果發現患者個子很高,手指細長,有近視,顎弓較高就要考慮到馬凡綜合徵,給他/她開主動脈CTA、心臟超聲等檢查。
另一方面,現在病人對馬凡綜合徵的認識也在逐漸提高,起碼在教育更發達的沿海地區,大家的認識提高很多。比如很多人在確診馬凡綜合徵後,很快會想到自己的孩子將來是不是也會得這個病,那麼就可以及早發現和治療。
不能一次治癒,及早發現和干預很重要
澎湃科技:也就是說,這不是一個絕症,早期進行干預的話是可以控製的?
王哲:對。這不是腫瘤,把病人有問題的血管換掉了以後,危及生命的問題就好了。但是馬凡綜合徵麻煩的地方在於,它是全身性的,一般不可能一次把病人全身有問題的血管都換掉,或者一次把全身的病都治癒。醫生只能先對可能引發致命危險的地方進行處理,讓它不危及生命,比如換掉血管或者心臟瓣膜。其他不危及生命的問題,比如視網膜脫落,可能也需要動手術,但根據病情的發展和嚴重程度進行就可以。目前也還沒有基因治療的方式可以治癒它。
澎湃科技:目前治療方式上有什麼共識嗎?
王哲:主要是對症治療,對於不危及生命的問題,如眼睛、關節等,等到問題出現時進行相應的治療即可。主要還是考慮心血管的問題。在早期,一般是通過藥物治療延緩病情,目前我們使用倍他樂克加一個血管緊張素受體拮抗劑(ARB)類或者血管緊張素轉換酶抑製劑(ACEI)類的降壓藥,有利於減緩血管的擴張。如果能儘早發現,可以每年或每半年進行一次隨訪,預防出現主動脈夾層等問題。在疾病進展的過程中,如果出現心臟的問題,可以選擇合適的時間介入,有利於減小手術風險和改善預後。
患者升主動脈直徑到5公分以上,就需要進行手術,因為發生夾層及破裂的幾率明顯上升。其實血管擴張到4-5公分之間,也有破裂的可能性,只是幾率相對較低。升主動脈一旦變成主動脈夾層,手術的難度及手術的風險會明顯增大很多,病人的預後也差很多。具體來說,如果患者只是升主動脈擴張,換一段人造血管就可以了,如果升主動脈根部及主動脈瓣有問題,根據患者的情況可以做瓣膜成型術(David手術)或做瓣膜置換術(Bentall手術)。但如果變成主動脈夾層,就要替換主動脈弓、升主動脈,還會影響到降主動脈,影響患者的預後。
馬凡綜合徵患者如果出現主動脈夾層,和其他主動脈夾層患者的情況還不完全一樣。中國的主動脈夾層主要是高血壓和動脈粥樣硬化引起的,這些患者替換升主動脈和主脈動弓之後,降主動脈膨大的速度比較慢,可能十幾年到幾十年才需要再次手術,置換胸降主動脈和腹主動脈。而馬凡綜合徵患者的動脈擴張進展比較快,升主動脈、主動脈弓因夾層急診替換後,胸降主動脈和腹主動脈擴張的速度比一般夾層患者要快,數年或十數年後就需要再次手術了。
澎湃科技:馬凡綜合徵患者的疾病負擔怎麼樣?
王哲:我覺得是比較重的,因為他們可能要做很多手術。例如心臟出問題之後,大部分情況下也不是一次手術就能解決的,經常需要2-3次手術。如果不算醫保的話,一次大型心臟手術大約需要10萬元,如果算上醫保的報銷,可能需要2-3萬元。對於一個家庭來說,可能是一個比較大的負擔。
對患者個體來說,因為他們個子高,身材相對纖長,在年輕的時候會比較出眾,各方面也表現有一定優勢,但突然被發現患有馬凡綜合徵後,心理上可能一下子無法接受,表現出一定的沮喪,我們也經常和他們解釋很多,大部分患者在充分瞭解以後還是可以接受的。另一方面,做完一次手術,患者可能會不時想到還有下次手術、再下次手術,影響到他們的工作和生活,所以患者的心理治療也很重要。
澎湃科技:你在臨床上有沒有接觸到印象比較深刻的馬凡綜合徵患者或家庭?
王哲:大約10年前,我碰到過一個馬凡綜合徵患者,來就診的時候,他的升主動脈已經擴張到9公分了,他運氣比較好,還沒有破。這對患者的影響很大,心臟功能下降,瓣膜也出現問題,算是在鬼門關繞了一圈了。對於醫生來說,手術的困難也很大,我們花了很大的力氣才把他救回來。這也是我強調要早發現、早診斷、早治療的原因。
這位患者之前有胸悶不適等症狀,但可能因為忍耐力比較強,一直沒有就診。所以我也要提醒大家,主動脈夾層的一個重要症狀就是劇烈的胸痛、背痛,像刀割一樣的疼痛,還有可能是遊走性的,如果出現這種情況,建議立刻就診,提早一小時都可能是救命的。
去年,他的兒子也確診了馬凡綜合徵,現在已經開始隨訪了,這對他的家庭來說是很大的影響。如果一個人被發現患有馬凡綜合徵,應該在他的家族中進行馬凡綜合徵的篩查,因為遺傳的概率有50%,還是很高的,除了父母和孩子,舅舅、叔叔等也有可能患有馬凡綜合徵。